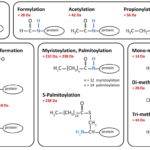
Protein Protein Conjugation Techniques and Methods
What is Protein-Protein Conjugation and How Is It Done? Conjugation techniques depend on two interrelated chemistries: functional groups present on…
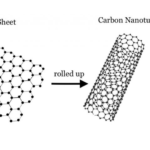
Bioconjugation of Carbon Nanotubes – Overview and Methods
We discuss different methods for the bioconjugation of carbon nanotubes including amine, aldehyde and carboxyl based methods.
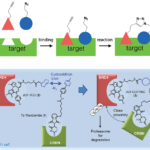
Orthogonal Bioconjugation Techniques – Step by Step Methods, and Impact
We discuss orthogonal bioconjugation techniques and methods such as SPAAC & tetrazine ligation, as well as applications of bioorthogonal chemistry.
Bioconjugation to Surfaces – Methods and Theory
We discuss different methods for bioconjugation to surfaces of polymers, DNA, and proteins such as tyrosine and lysine bioconjugation.
Methionine Selective Bioconjugation Methods and Theory
We discuss different methionine selective bioconjugation techniques utilized for protein antibody, polymer, or surface conjugation with step-by-step instructions.
Methods for Protein and Antibody Bioconjugation to Gold
We discuss methods for protein and antibody bioconjugation to gold including theory, alternative approaches, and protocols.
Protein Labeling with Fluorescent Probes – Theory and Methods
We discuss types of fluorescent probes, how to select a fluorescent probe for protein labeling, protein labeling kits, and protocols…
Protein Conjugation Chemistry – Theory and Examples
We discuss protein conjugation chemistry techniques such as lysine, cystein, or site-specific conjugation using bifunctional linkers.
Biopolymer surface functionalization: Simple Step-By-Step guide
Introduction to Surface Functionalization of Biopolymers Immobilizing (or covalently attaching) proteins, lipids, carbohydrates, and other polymers on biopolymer surfaces is…
Tips For Making Research Efficient
Insights from Priyamvada Jayaprakash. Connect at Her LinkedIn Profile. She’d love to discuss her PhD experience with you and get…