Lately, scientists in fields such as chemical biology, pharmacology, and medicinal biochemistry have shown interest in developing methods to study biomolecules in live cells. These novel techniques are known as Orthogonal Bioconjugation techniques, or simply, bioorthogonal reactions. They are required to figure out biological processes at a relevant time scale without having to add a high concentration of toxic reagents.
Orthogonal Bioconjugation techniques have gained significance among scientists because they allow biomolecules to be conjugated specifically and quickly by utilizing naturally occurring functional groups.
A few examples of Orthogonal Bioconjugation techniques are: bioorthogonal click chemistry, tetrazine ligation, Staudinger ligation, Cu-free click chemistry (SPAAC), click reaction and photo-click reaction.
These are also the most preferred techniques for labeling biomolecules within individual cells as well as entire organisms. Bioorthogonal Chemistry has triggered a colossal shift in the reaction site in the field of chemical biology. An aqueous mixture of proteins and sugars has displaced the refluxing of toluene, while reactions have been adapted to be performed inside a living cell. Traditional round bottom reaction vessels are now being replaced by biological species such as cancer cells and zebrafish.

What is Orthogonal Chemistry?
Orthogonal chemistry refers to that arena in chemistry involving a special kind of reaction between two pairs of substances where each member of a pair interacts selectively with its respective partner, but is not prone to interacting with either member of the other pair.
Bioorthogonal reactions are a specific type of chemical interaction that can occur within living organisms without interfering with native biochemical processes. The most distinct features of these reactions include fast kinetics, tolerance to an aqueous environment, high selectivity, as well as compatibility with naturally occurring functional groups. Bioorthogonal chemistry was first coined by Carolyn R. Bertozzi in 2003 who recently won The Solvay Prize 2020 for her discovery of the field.

Biomolecular study within living cells is often complicated by the complexity of biological systems; thus overcoming this obstacle has long been a subject of research. As a result, a bioorthogonal technique has been developed to selectively tag the biomolecule of interest with a fluorophore. Once tagged, the desired biomolecule gives off a unique signal that allows it to be easily identified and studied within the complex biological setting. The process is done via two steps:
Step 1. Incorporation of Metabolite with a Functional Group
As shown in the schematic diagram above, cells are first incubated with a metabolic precursor having a unique functional group which is known as the Chemical Reporter. This metabolite(red in the diagram) could be a monosaccharide for glycan labeling, a nucleotide for DNA labeling, an amino acid for protein labeling, or a fatty acid for lipid labeling.
We’ve discussed more about methionine selective bioconjugation and protein conjugation chemistry in our other articles.
Step 2. Treating with Bioorthogonal Reagents
Once the chemical reporter is incorporated into the target biomolecule, the next task is to treat the target with a probe molecule that bears complementary bioorthogonal functionality. Ideally, bioorthogonal reagents should contain a reactive entity that will solely react with another specific reactive group leaving no potential for cross-reactivity with any other biomolecule functional groups.
In other words, a bioorthogonal reactive group could be added to a complex mixture of biological molecules in an aqueous solution without interfering with any of them. This ‘biological inertness’ is the major property of any bioorthogonal reagent. A number of bioorthogonal functional groups are currently available, each with its unique advantages as well as shortcomings.
For more information about protein labeling with fluorescent probes, take a look at our article.
Common Orthogonal Bioconjugation Techniques
Some of the most common techniques in Orthogonal Bioconjugation include bioorthogonal click chemistry (CuAAC), tetrazine ligation, Staudinger ligation and Cu-free click chemistry (SPAAC).
These techniques have brought about radical changes to the conventional practices hitherto followed in protein bioconjugation.
1. Bioorthogonal Click Chemistry
Click chemistry, or Cu-catalyzed Azide-Alkyne Cycloaddition (CuAAC), is based on how enzymes utilize transition metals to catalyze biochemical reactions within living cells. This technique has been the center of focus to bioorthogonal chemistry for much of the last decade.
Copper-catalyzed Azide-Alkyne 1, 3-dipolar cycloaddition carries most of the features of an ideal click reaction such as selectivity, efficiency, and simplicity. Triazoles, generated from CuAAC, have unique properties of stability and chemoselectivity. They can remain stable under different conditions, such as acidic or basic hydrolysis, oxidation, and reduction.
Due to the high dipole moment of heterocycles, triazoles can easily contribute to the formation of hydrogen bonding and dipole-dipole interactions. However, the reactive groups of azide and alkyne involved in CuAAC have beneficial attributes like their small size, stability, and inactivity under physiological conditions, which consequently qualify them for labeling proteins.
A high atom efficiency, inherent selectivity, and tunable electronics led to the idea of introducing dipolar cycloaddition reactions to biomolecules. In addition, the inertness to irrelevant functional groups in their native settings demonstrates the intriguing potential of this type of chemistry for bioconjugation. Also
1,3-dipolar cycloadditions are significantly enhanced in aqueous media; this is precisely why this reaction is the perfect candidate for the tethering of biomolecules.
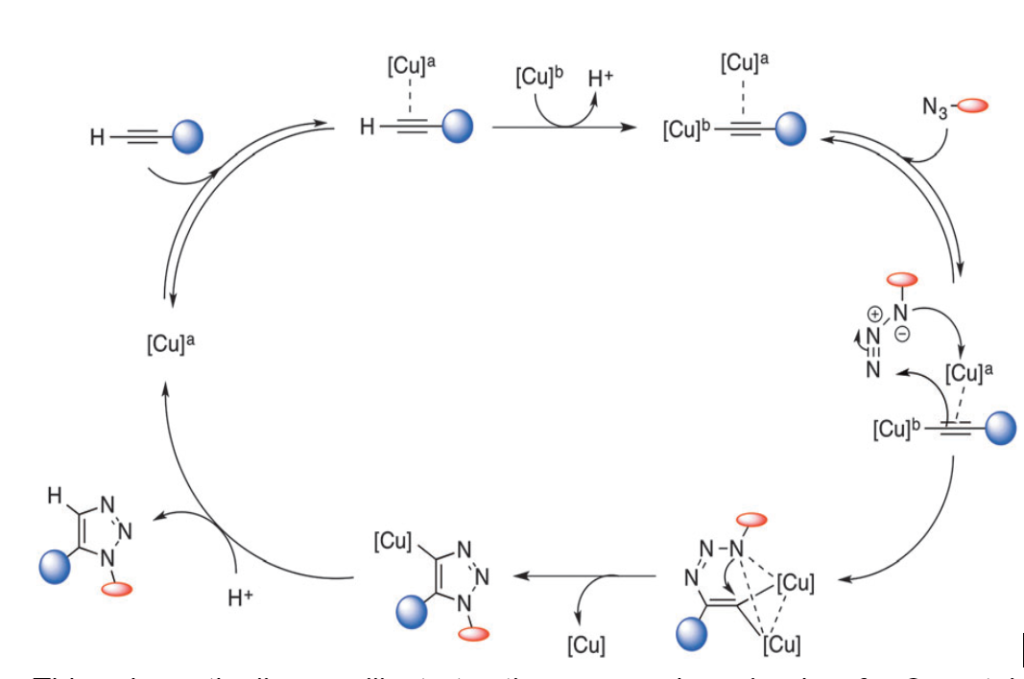
However, the toxicity of Cu(I) lowers the biocompatibility level of CuAAC reactions and impedes their application in living cells.
Several ligands have recently been developed to counteract this issue. These can increase the reactivity of Cu(I); the fewer Cu(I) ions are available, the lower the toxicity. In addition, CuAAC can be accelerated by the use of ligands with stabilizing effects such as tris[(1-benzyl-1H-1,2,3-triazol-4-yl)- methyl]amine (TBTA) and tris(3-hydroxypropyl -triazolyl methyl)- amine (THPTA).
As a matter of fact, the utilization of ligands has broadened the biological applications of CuAAC, for instance, allowing live-cell imaging. Likewise, water-soluble THPTA ligand was successfully employed for the CuAAC-mediated functionalization of the cowpea mosaic virus; this was previously attained via using a sulfonated bathophenanthroline (BPS) ligand.

We’ve discussed more protein conjugation chemistry techniques in our related article.
2. Tetrazine Ligation Reactions and Mechanisms
The interaction between a trans-cyclooctene and an s-tetrazine in an inverse-demand Diels Alder reaction followed by a retro-Diels Alder reaction is functionally known as the Tetrazine Ligation reaction.
Tetrazine, composed of a six-membered aromatic ring with four nitrogen atoms, is a product of the cycloaddition reaction between tetrazine and alkene. While its ligation chemistry was first reported by Sauer in the 1990s, bioorthogonal tetrazine ligation was independently reported by Fox and Hilderbrand in 2008. They used trans-cyclooctene and norbornene respectively to react with tetrazine in aqueous solution.
Tetrazine ligation is observed to proceed extremely fast, with a second-order rate constant as high as 2000 M−1s−1. Among several tetrazine isomers, 1,2,4,5-tetrazine is employed most often in tetrazine ligation. An instance of a reaction of 1,2,4,5-tetrazine with strained alkenes or alkynes uses an “inverse electron-demand Diels–Alder reaction” (iEDDA), in which dienes (tetrazines) are electron-deficient due to electron-withdrawing substituents, whereas dienophiles (alkenes or alkynes) are electron-rich due to electron-donating substituents.
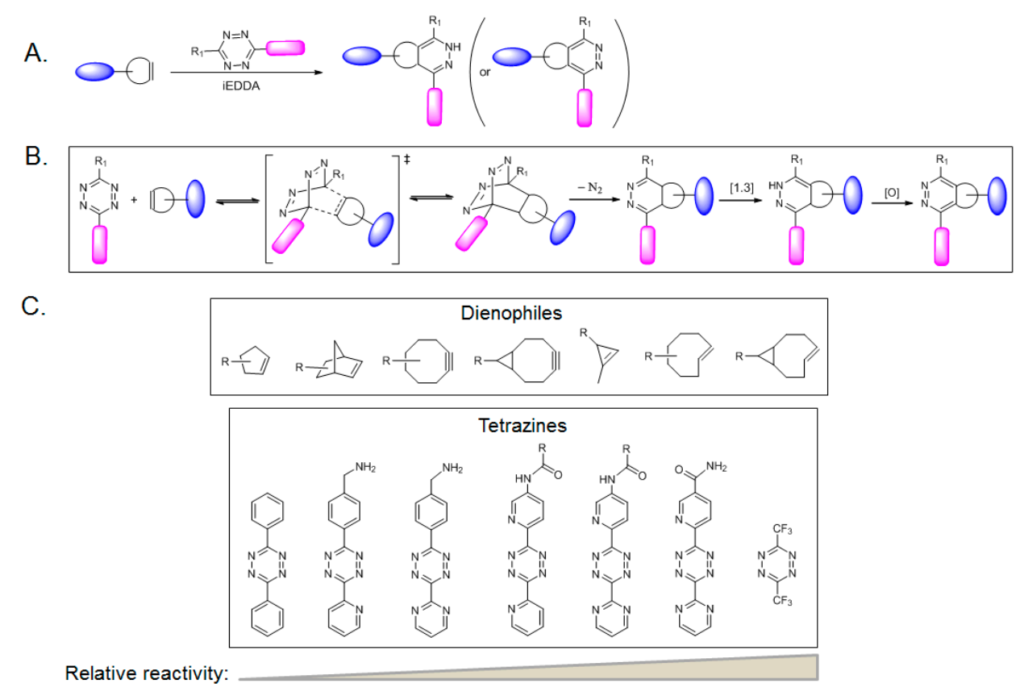
The iEDDA reaction rate for this reaction is extraordinarily high. Moreover, a fluorescence-quenching mechanism can be used in the tetrazine ligation through iEDDA. The fluorophore-conjugated tetrazine reagents also show the ‘‘turn-on’’ fluorescence phenomenon. Tetrazine quenches the fluorescence of a covalently linked fluorophore; upon cycloaddition with an alkene, the fluorescence is recovered due to the loss of the tetrazine moiety.

A few drawbacks of Tetrazines are its relatively high cost and availability of precursors, which has limited their synthesis to a small scale. Tetrazines are also held back due to their high reactivity, particularly those with high nitrogen content. This is why they are mostly synthesized in a well-ventilated hood.
On the other hand, their fast reaction rates, bioorthogonality, and mutual orthogonality with other click reactions have popularized this chemistry to be utilized in a wide variety of biochemical research.
3. Staudinger Ligation Reaction
The stepwise synthesis of amines starting with triphenylphosphine and azides is known as the Staudinger Reaction, named after Nobel Prize-winning chemist, Hermann Staudinger early in the last century.
(Staudinger and Meyer, 1919).
Here, Triphenylphosphine reacts with azides to form an intermediate iminophosphorane with the release of nitrogen gas. This intermediate quickly breaks down in aqueous environments to produce triphenylphosphine oxide and a primary amine. This reaction has since been successfully employed to synthesize amines. Often azides are thought of as hidden amines because the azide is relatively inert to other reactants until it is revealed through the Staudinger reaction.

Ortho positioning of an electrophilic benzyl methyl ester group on one of the phenyl rings provides a reactive site for the nucleophilic nitrogen from an azide group; this temporarily forms an aza-ylide interaction with the phosphorus atom core. Nucleophilic attack of the nitrogen on the carbonyl “electrophilic trap” releases methanol and forms a stable amide bond.

4. Orthogonal Copper-free Click Chemistry, SPAAC reaction
Strain-Promoted Alkyne-Azide Cycloaddition (SPAAC), also known as Cu-free click reaction refers to the rapid cycloaddition between a strained cycloalkyne and azide. The strain in the alkyne makes it highly reactive under physiological conditions, eliminating the need for the toxic copper catalyst.
Although this reaction between azides and cyclooctynes had already been discovered nearly 65 years earlier in the year 1953 by Blomquist et al, it had never been deployed in living systems. The benefit of this reaction on live cells was first demonstrated by Agard et al. He metabolically labeled Chinese hamster ovary (CHO) cells in culture with Ac4ManNAz followed by reaction with a cyclooctyne-based molecule.
Bertozzi worked upon the possible substitutions on cyclooctene and found difluorinated cyclooctyne showed higher reactivity. Later, Boons developed biarylazacyclooctynone by fusing two benzene rings to cyclooctyne, creating a second-order rate constant which was approximately three times higher than that of the simple cyclooctyne.
Meanwhile, Van Delft raised the reactivity of simple cyclooctyne by introducing an amide bond into the ring, which could easily be synthesized in high yields. He further developed bicyclononyne as another cyclooctyne analog for click chemistry, which also exhibited high reactivity towards azide.

Since its rediscovery, research focus has been shifted towards redesigning of the cyclooctyne scaffold to improve some important features of SPAAC, including reaction rate, solubility of cyclooctyne reagents, as well as undesired reactivity. For instance, Baskin et al tuned the electronics by the addition of electron-withdrawing fluorine atoms to yield monofluorocyclooctyne (MOFO) and difluorinated cyclooctyne (DIFO) scaffolds with an increased rate.
The addition of ring strain has produced dibenzocyclooctyne (interchangeably called DIBO or DBCO) with higher reactivity. Moreover, the inclusion of scaffolds bearing stabilizing amide groups has resulted in aza dibenzocyclooctyne (ADIBOs) and biarylaza cyclooctyne (BARAC), striking an excellent balance between stability and reactivity.
Altering the cyclooctyne scaffold has generated a series of cyclooctynes that can be utilized based on the desired application.
The most recent development in strained alkynes was reported by Bertozzi and co-workers wherein they replaced two methylene groups with a sulfur atom to yield 3,3,6,6-tetramethyl thiacycloheptyne (TMTH).
The use of TMTH as a bioorthogonal labeling reagent has been demonstrated through cell surface modification of azido-functionalized glycans as well as selective labeling of barstar protein-encoding azidohomoalanine.

Recently, a new cell labeling method, with imageable probes, has been discovered involving the application of metabolic glycoengineering and Cu-free click chemistry. The combination of metabolic glycoengineering and Cu-free click chemistry allows for the stable labeling of cells with various molecules without affecting the characteristics of the cells. Therefore, this method is expected to overcome the problems associated with the direct cell labeling method.

We’ve discussed other methods for biopolymer surface functionalization in our related article.
Orthogonal Bioconjugation Protocol Example – Studying Cellular processes utilizing SPAAC protocols
A large number of SPAAC protocols are available for studying various cellular processes. The dynamic modification of intracellular proteins by O-linked β-N-acetylglucosamine (O’GlcNAcylation) plays a critical role in many cellular processes. Although there are various methods for O-GlcNAc detection, there are only a few techniques for monitoring glycosylation stoichiometry and state. The required materials and stepwise procedure of a SPAAC-based approach for the determination of OGlcNAc stoichiometry and state, is shown below, based on a publication of the American Chemical Society.
Materials Required
- 0.05% SDS buffer [0.05% SDS, 150 mM NaCl, and 10 mM TEA (pH 7.4)]
- 4% SDS buffer [4% SDS, 150 mM NaCl, and 10 mM TEA (pH 7.4)]
- 1% SDS buffer [1% SDS, 150 mM NaCl, and 10 mM TEA (pH 7.4)]
- 1% SDS GalT buffer [1% SDS with 20 mM HEPES (pH7.9)]
- UDP-GalNAz [0.5 mM in 10 mM HEPES (pH 7.9)]
- Iodoacetamide (made fresh, 600 mM stock in H2O)
- DBCO-PEG-5K (10 mM stock in DMSO)
- 2× loading buffer [20% glycerol, 0.2% bromophenol blue, and 1.4% β-mercaptoethanol (pH 6.8)]
- Complete, Mini, EDTA-free Protease Inhibitor Cocktail
- Benzonase
- Control antibodies for Western blotting: anti-CREB1
Step-by-step protocol:
- Cells are collected and washed twice with PBS (1 cm×10 cm dish at ≥80% confluency).
- 26 μL of H2O, which contains 10× Complete Mini Protease Inhibitor Cocktail, is added.
- A 50 μL of 0.05% SDS buffer is added followed by resuspending the cells.
- 1 μL of benzonase is added and incubated on ice for 30 min, followed by the addition of 200 μL of 4% SDS buffer.
- Any debris is pelleted by centrifugation at 10000g for 10 min at 15°C, preceded by vortex-mixing
- Perform the BCA assay and normalize the protein concentration to 1 μg/μL using 1% SDS buffer.
- To 1 mg of protein (1000 μL),
i) 3000 μL of methanol is added and vortex-mixed
ii) 750 μL of chloroform is added and vortex-mixed.
iii) 2000 μL of H2O is added and vortex-mixed.
iv) The resulting mixture is then centrifuged at 5000g for 5 min at 15°C.
v) The upper aqueous phase is discarded, while leaving the interface layer interacting.
vi) 2250 μL of methanol is added and vortex-mixed.
vii) The solution is then centrifuged at 5000g for 10 min at 15°C and the supernatant is discarded.
viii) The pellet is then allowed to air-dry for 5 min.
[ Note: The protein pellet should not be allowed to “overdry”, as this will make the proteins difficult to resuspend, which is true for all subsequent drying steps.]
- The protein is resuspended in 100 μL of 1% SDS GalT buffer.
- Let it be sonicated until the proteins are dissolved.
- The BCA assay is performed and the protein is normalized to a concentration to 2.5 μg/μL using 1% SDS GalT buffer.
- The enzymatic reagents are added in the following order for the “+GalT + UDP sample”:
i) 40 μL of lysate
ii) 49 μL of H2O
iii) 80 μL of labeling buffer
iv) 11 μL of MnCl2
v) 10 μL of UDP-GalNAz
vi) 10 μL of GalT enzyme
- The enzymatic reagents are added in the following order for the “-GalT + UDP sample”:
i) 40 μL of lysate
ii) 59 μL of H2O
iii) 80 μL of labeling buffer
iv) 11 μL of MnCl2
v) 10 μL of UDP-GalNAz
- Optional: The enzymatic reagents are added in the following order for the “+GalT – UDP sample”
i) 40 μL of lysate
ii) 59 μL of H2O
iii) 80 μL of labeling buffer
iv) 11 μL of MnCl2.
v) 10 μL of GalT enzyme
- The samples are incubated at 4°C for 20 hours.
- 7.5 μL of freshly made iodoacetamide (600 mM) is added to each sample.
- The resulting solution is further incubated for 30 min in the dark.
- To each sample:
i) 622 μL of methanol is added and vortex-mixed.
ii) 156 μL of chloroform is added and vortex-mixed.
iii) 415 μL of H2O is added and vortex-mixed.
iv) The solution is centrifuged at 10000g for 5 min at 15°C.
v) Discard The upper aqueous phase is discarded while leaving the interface layer is added.
vi) 467 μL of methanol is added into the tube to the interface layer and vortex-mixed.
vii) The solution is then centrifuged at 10000g for 10 min at 15°C and the supernatant is discarded.
viii) The pellet is allowed to air-dry for 5 min.
ix) 90 μL of 1% SDS is added followed by sonication until the proteins are dissolved.
- 10 μL of DBCO-PEG-5K is added and vortex-mixed.
- The mixture is then incubated for 16 h at room temperature or boiled for 5 min at 98°C.
- To each sample:
i) 300 μL of methanol is added and vortex-mixed.
ii) 75 μL of chloroform is added and vortex-mixed.
iii) 200 μL of H2O is added and vortex-mixed.
iv) The solution is centrifuged at 10000g for 5 min at 15°C.
v) The upper aqueous phase is discarded while leaving the interface layer intact.
vi) 225 μL of methanol is added into the tube to the interface layer and
vortex-mixed.
vii) The resulting solution is centrifuged again at 10000g for 10 min at 15°C and the supernatant is discarded.
viii) The pellets are allowed to air-dry for 5 min.
- 25 μL of 4% SDS is added followed by sonication until proteins are dissolved.
- 25 μL of 2× SDS is added followed by boiling for 5 min at 98°C.
- About 40 μg per lane is loaded on SDS-PAGE for Western blotting.
Applications of Bioorthogonal Chemistry in Science
The numerous applications of bioorthogonal chemistry range from biochemical studies and design of tools for the ligation of large biomolecules, to advances in the ability to track small molecular changes such as post translational modifications.
For instance, one application of bioorthogonal chemistry helps to improve our understanding of glycosylation in animal cells. Another application involves the attachment of functional groups to therapeutically relevant proteins such as antibodies. Bioorthogonal reactions have also made it possible to assemble molecular imaging agents within living tissue, in order to detect disease.
The impact of bioorthogonal chemistry is evident in its research and therapeutic applications. Ranging from synthetic biochemistry to polymer sciences and drug discovery; from glycan engineering to in vivo imaging and beyond. It has opened up new avenues for biological investigation and produced outstanding discoveries in areas as diverse as protein biophysics, neurophysiology, developmental and stem cell biology, and cancer detection. Also, as researchers continually work on improving the field of bioorthogonal chemistry, they have found these same reaction mechanisms useful in their research.
Applications of Orthogonal Bioconjugation Techniques
Application 1. Chemical Reporters and Their Bioorthogonal Reactions for Labeling Protein O-GlcNAcylation
Protein O-GlcNAcylation refers to the unconventional glycosylation of nuclear, mitochondrial, and cytoplasmic proteins by attaching a single O-linked β-N-acetyl-glucosamine (O-GlcNAc) moiety. Advancements in the labeling and identification of O-GlcNAcylated proteins have provided deeper insight into the basic molecular biology of O-GlcNAcylation, as well as the roles of cell signaling and gene regulation in physiology and disease.
To label a protein with O-GlcNAc, a Metabolic Chemical Reporter (MCR) needs to be utilized as shown in the diagram below. MCRs have unique reactivity and can be introduced into the naturally occurring biomolecules of living tissue, through the biosynthetic machinery of the cell.
Using bioorthogonal labeling reactions, these MCRs are reacted with specifically designed molecular probes. This process allows biomolecules of interest to be visualized and isolated. To ensure a successful reaction, MCR groups must be non-toxic to the cell and chemically stable prior to the reaction. Also, the MCR groups are to react solely with the probes and neither of them should interact with any native biomolecules of the cell.

Application 2. Bioorthogonal Metabolic Labeling of Viruses
Bioorthogonal metabolic labeling, via the typical cellular metabolic pathways (e.g., phospholipid and sugar), is a promising method for the efficient labeling of live viruses. Nonenveloped viruses are usually difficult to label due to a lack of host-derived envelopes. However, with a view to labeling and tracing nonenveloped viruses, a novel bioorthogonal labeling strategy is being developed utilizing the protein synthesis pathway.
Azido motifs are first introduced into the viral capsid proteins by substituting methionine residues during viral protein biosynthesis and assembly. Afterwards, the azide-modified EV71 (N3-EV71) particles are labeled with dibenzocyclooctyne (DBCO)-functionalized fluorescence probes through a bioorthogonal reaction that does not affect viral infectivity.
Dual-labeled imaging reveals that EN71 virions primarily bind to scavenger receptors and are absorbed by the cell through clathrin-mediated endocytosis. Afterwards, the viral particles are transported into early and late endosomes where viral RNA is released in a low-pH dependent system, approximately 70 mins after infection. The results clarify the mechanisms for viral trafficking and uncoating, which may shed light on the pathogenesis of EV71 infection and contribute to antiviral drug discovery.

Application 3. Labeling and Visualizing Tumors
Cancer nanomedicine, particularly tumor-specific targeting and imaging, is an application of bioorthogonal chemistry that has created a lot of excitement among researchers. Bioorthogonal click chemistry is the technique that has overcome the challenges of conventional active-targeting strategies such as heterogeneous expressions of tumor surface receptors and the slow pharmacokinetics of targeting probes.
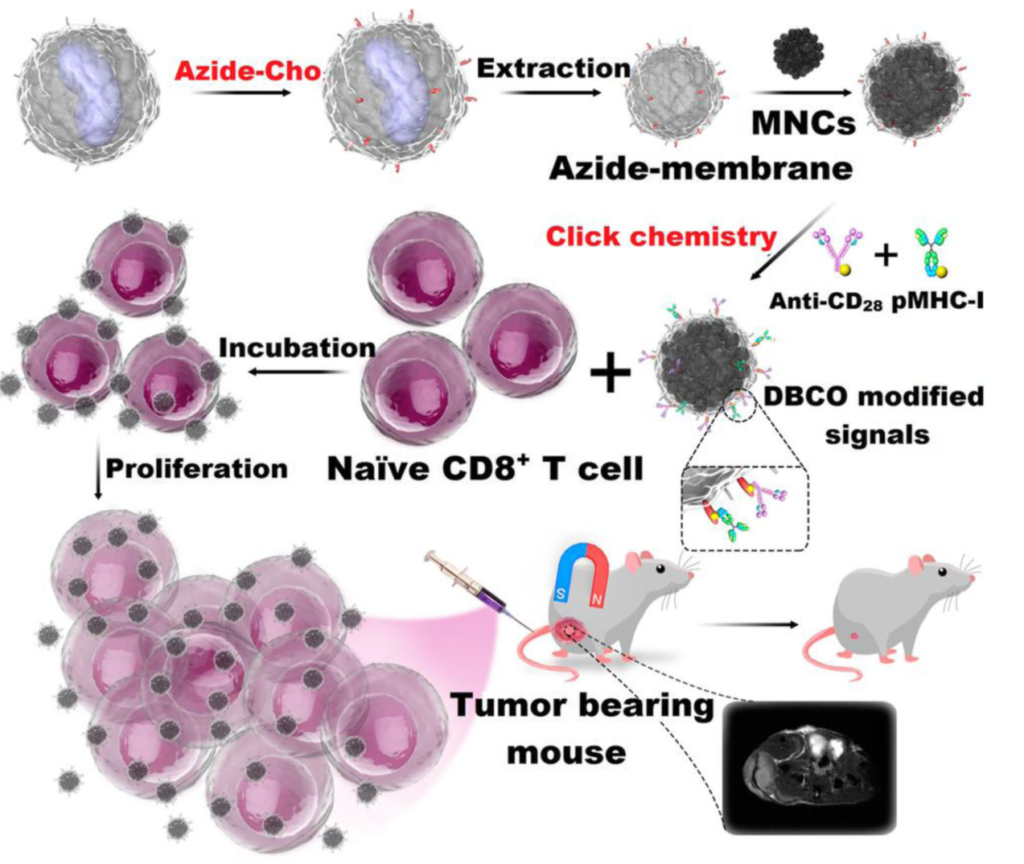
Using Cu-free click chemistry, in 2017, Zhang et al. modified their biomimetic magnetosomes based on artificial antigen-presenting cells (aAPCs) with stimulatory signals. Azide groups were introduced on the surface of leucocyte membranes via azide-choline biosynthesis. This made it possible for the inner components of the leucocytes to be extracted.
Next, the aAPCs were fitted with dibenzocyclooctyne (DBCO)-modified T-cell stimulatory signals using click chemistry with azide groups. Stimulatory signals such as pMHC-I and anti-CD28 were then localized on aAPCs to stimulate T cells.
T-cell-based therapy depends on two major factors: targeting performance within the tissues, and T-cell expansion and stimulation outside the tissues. Synthesized aAPCs with T-cell stimuli showed a high affinity for Cytotoxic T-cells (CTLs). Thus, they were effective for in-vitro expansion of antigen-specific CTLs.
Within a living system, they will likely hit more CTLs at the tumor sites. The magnetic nanoparticles attached to the aAPCs made it possible to monitor aAPC-CTL complexes via MR imaging using T2 contrast signal intensity. The murine lymphoma model experiment showed that aAPCs were efficient in adaptive T-cell based anticancer immunotherapy within the living system.
Application 4. Targeted Delivery Of Nanoparticles
In 2012, an interesting tumor-targeting strategy was introduced by Koo et al for the targeted delivery of nanoparticles based on metabolic glycoengineering and an orthogonal bioconjugation technique known as click chemistry.
Using tetraacetylated N- azidoacetyl -d-mannosamine (Ac4ManNAz) as a precursor, the targetable azide group containing glycans was artificially introduced to the tumor cells. This was followed by treating the cells with DBCO-conjugated liposomes (DBCO-lipo). These DBCO groups were found to bind strongly and specifically to azide groups on the surface of target cancer cells using a method known as Cu-free click chemistry.
While in vitro study showed that the cellular uptake of DBCO-lipo was relatively higher than the control PEG-lipo; in vivo study demonstrated that the incorporation of azide groups on tumor tissue could be controlled by metabolic glycoengineering if a dose-dependent means was used.
The tumor-targeting of DBCO-lipo was accelerated with the intratumoral injection of Ac4ManNAz and intravenous injection of DBCO-lipo.
When glycol chitosan nanoparticles(CNP) was impregnated with Ac4ManNAz and then injected into the tumor-bearing mice, the resulting Ac4ManNAz–CNPs were delivered to the tumor via EPR effect.
Afterwards, azide groups were generated on the tumor cells by metabolic glycoengineering. The next step was to intravenously inject BCN-modified and chlorin e6 (Ce6)-loaded CNPs into the same mice. Further click chemistry was used to conjugate the BCN groups on the surface of the BCN-Ce6-CNPs to the azide groups generated on the tumor surface. This click reaction enhanced the accumulation of BCN-Ce6-CNPs in tumor tissue, creating an efficient strategy for drug delivery and tumor therapy.

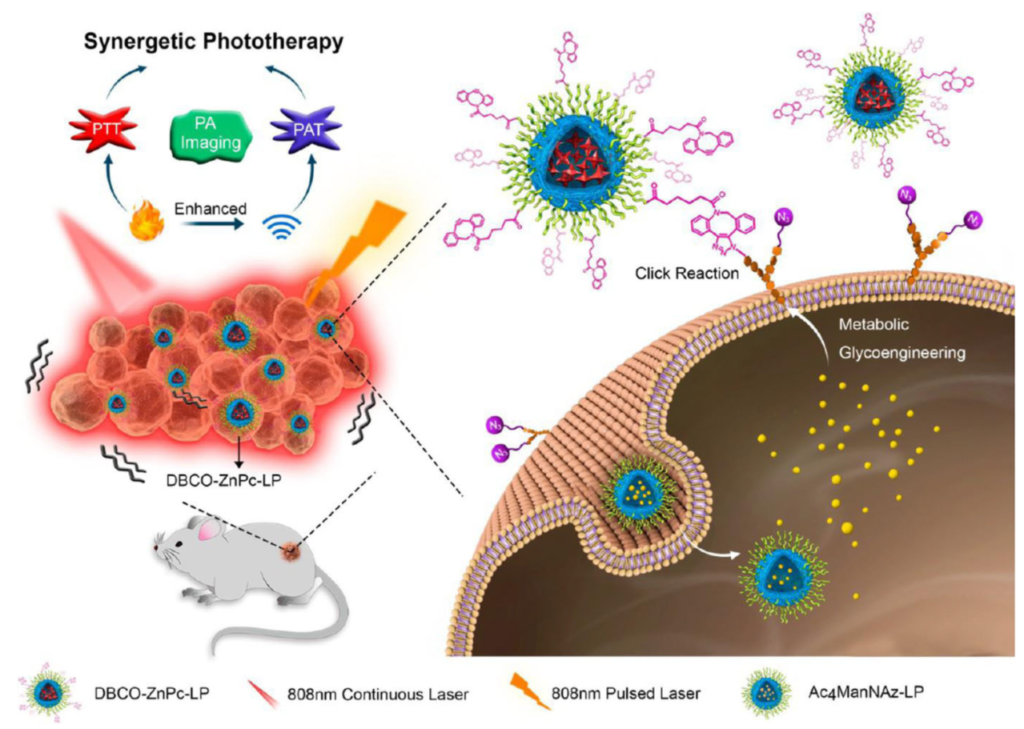
More research based on Click Chemistry followed. In 2017, the work of Du L et al. was published. They had worked on in-vivo multifunctional nanoplatforms for photothermal therapy (PTT) and photoacoustic therapy (PAT). They developed DBCO-modified zinc (II)-phthalocyanine-loaded liposome (DBCO-ZnPc-LP) followed by a two-step tumor-targeting strategy via metabolic glycoengineering and click chemistry.
At first, a nude mouse was intravenously injected with Ac4Man-NAz-loaded liposomes, which can bind the azide group for a chemical receptor onto the cell surface via metabolic glycoengineering. The intravenous injection accelerated the accumulation of DBCO-ZnPc-LP in tumor tissue. The effect is similar to that of in-vivo Cu-free click chemistry on the reaction between azide groups on the cell surface and DBCO. However upon laser irradiation, the accumulated DBCO-ZnPc-LP was able to generate large amounts of heat and acoustic effects. The resulting PTT and PAT could efficiently destroy tumor tissue.
Future of Bioorthogonal Chemistry
Further steps in the field of bioorthogonal chemistry may lean towards pharmaceutical research, advancements in medical imaging, or even human applications as more reactions from the toolkit are tested preclinically.
Bioorthogonal reactions are innovative resources to better understand nature. They are the results of persistent employment of orthogonal chemistry within complex biological settings. Already, the idea of a bioorthogonal reaction has earned the credibility of the vast a majority of chemical biologists. This is reflected by the commercial availability of a large number of bioorthogonal reagents.

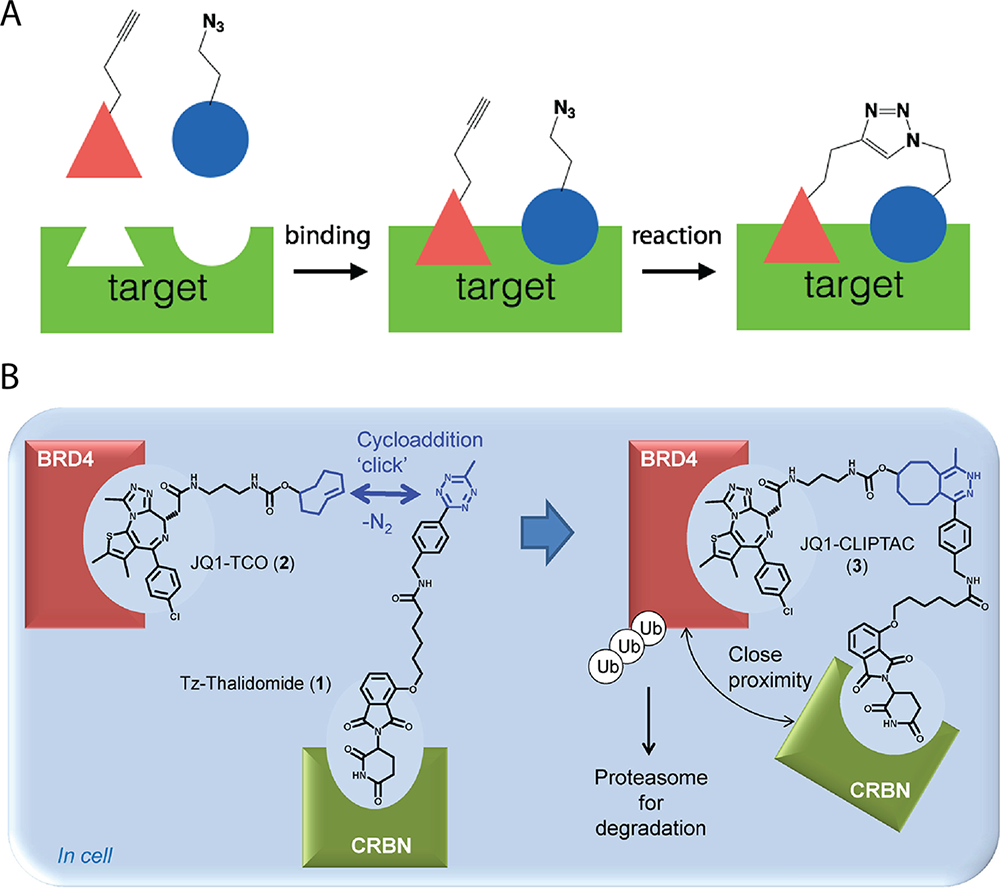
One potential application of bioorthogonal chemistry is in assembling drug fragments in situ, particularly for azide−alkyne cycloadditions. By designing bioorthogonal reactions between fragmengs of proteins that bind close to one another, protein targets can be used to catalyze the formation of their own inhibitors.
To illustrate, a target biomolecule such as an enzyme is compatible with two fragments bearing bioorthogonal functional groups. If the fragments are bound to the enzyme such that the functional groups come close to one another, a tighter binding compound may be formed. This method has been used for in situ discovery of enzyme inhibitors.
Researchers presume that such reactions can be designed to take place within living cells. They have deployed bioorthogonal reactions to form drug-like molecules inside cells.
Pre-targeted therapy (or imaging) using reactive monoclonal antibodies is another prospect of bioorthogonal chemistry. In case you’re interested, we’ve also discussed protein & antibody conjugation to gold in a related article.

Here, the target cells (blue) are exposed to an affinity ligand directly modified by an imaging or chemotherapeutic agent. Via a multi-step approach, the affinity ligand directly connected to trans-cyclooctene (a non-therapeutic and bioorthogonally reactive element) is delivered to the target cells. It is possible for a significant amount of toxic background signal to be released while binding to the target cells if there are excess long-circulating affinity ligands.
Current research applications and preclinical tests of bioorthogonal reactions are likely to be exhausted in the near future, leading to human applications. Although even the most established chemoselective reactions still need to be optimized, a few can be readily deployed, depending on the intended application.
As properties such as kinetics, stability, and reactive handle size are further optimized, new reactions will open up further therapeutic applications. Future studies are likely to lay greater stress on the application of bioorthogonal couplings to trigger the formation of biologically active components, whether by direct synthesis of drugs in living organisms or through triggered drug release. Exploring yet untapped organic reactions might lead to the evolution of even more bioorthogonal tools of chemistry. It is hoped that this expanded chemical toolbox will equip chemists and biologists to transmute these techniques into valuable biological applications.