Protein Purification Summary
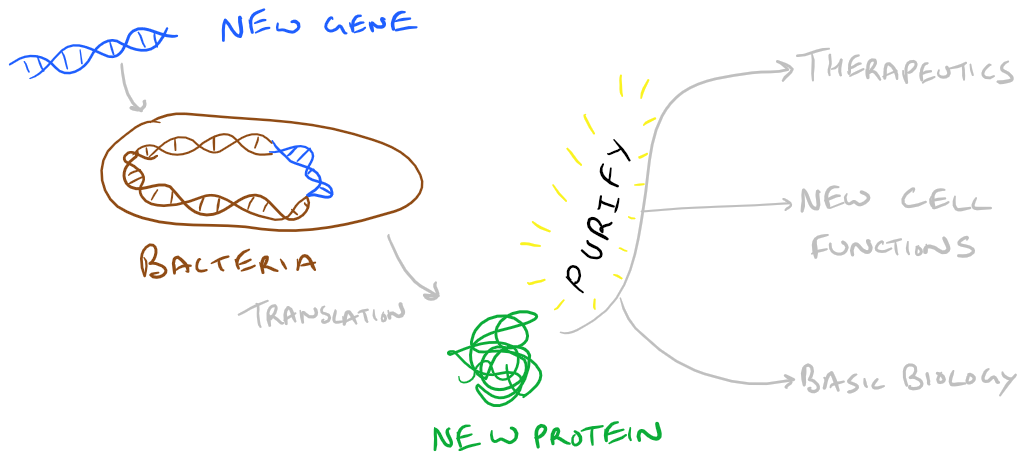
In our previous blog posts we have explored Gene cloning with Plasmid Vectors in Bacteria, Transient transfection into Mammalian Cells with Calcium Phosphate, and how we can use newly introduced proteins to control biology. Proteins made this way are considered recombinant because they aren’t natively produced in the organism that you got them from. We really like recombinant technology because it allows us to scale up protein production and generate therapeutic and/or interesting fusion proteins that we can use. If you want some human protein, would you rather grow humans and isolate the protein for scale up (~30 years per doubling)? Or use bacteria instead (~20 minutes per doubling)? Note: this was a joke. Don’t grow humans for protein production 🙂
In this blog post, we are going to explore how “recombinant” proteins can be purified after cells have expressed the gene products that you cloned into them. The strategies explored here can be applied to all sorts of proteins so let’s begin!
Note: You can easily troubleshoot your protein purification procedure by labeling your protein with a fluorescent probe. We’ve given you some more information in our article.
Strategies for Protein Purification
Let’s say you have some bacteria that you’ve produced a protein inside. Your first step is to lyse those bacteria and neutralize any proteases that are now in your lysate. Proteases will wreak havoc on all the proteins in solution…so this step is important. Next, we have to think about the recombinant protein that we created in order to purify it. Note that conjugated proteins can utilize their unique tags for purification.
Several different purification methods can be used based on your properties:
-
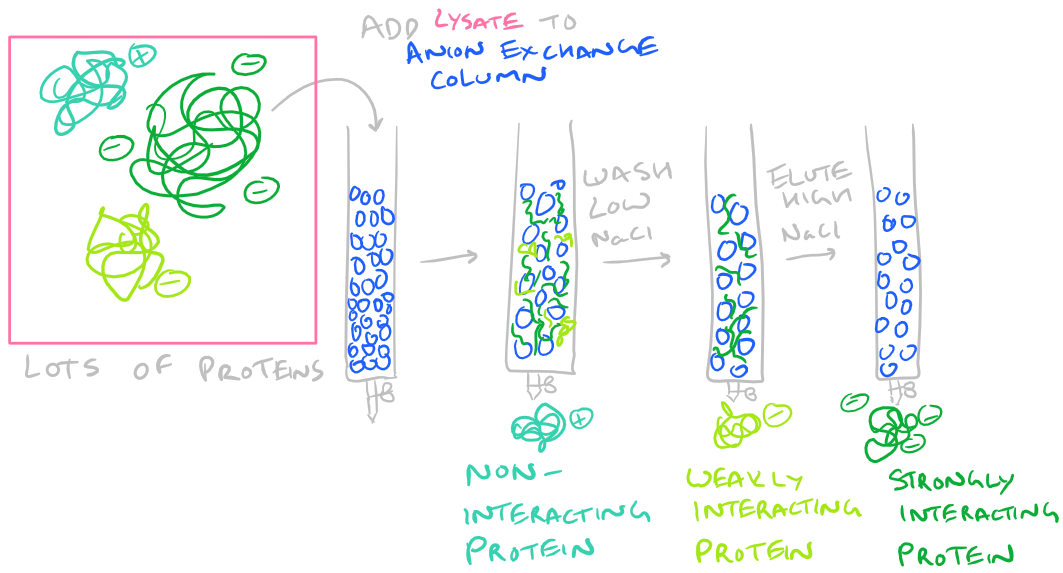
- Protein Charge: If your protein has a overall charge because of excess arginine or aspartamine residues, perhaps it can be purified by running it through an ion exchange column. For negatively charged proteins, use anion exchange chromatography, and for positively charged proteins use cation exchange chromatography. The steps here are simple…Dissolve your protein in a buffer and incubate it with the resin. Wash the resin with some low salt buffer. And then elute the bound protein with some high salt buffer (which breaks the ionic interactions with the resin).
-
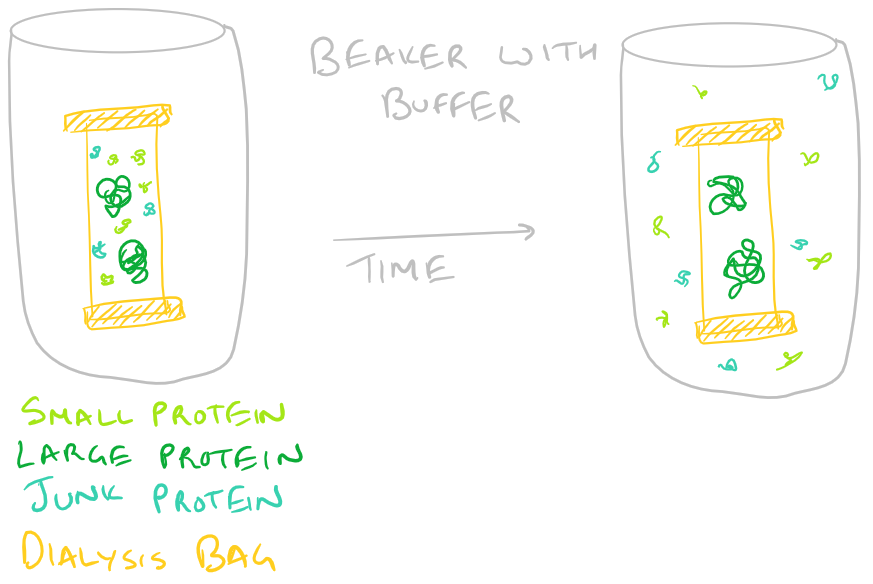
- Protein Size: Dialysis and Size Exclusion chromatography can help you isolate proteins based on their size. In the case of dialysis, you incubate your protein in a dialysis bag and stir it while replacing the buffer outside. Your protein and larger proteins are retained in the bag while smaller proteins are filtered out through diffusion. Size exclusion chromatography (SEC) works similarly to separate out larger molecules from smaller ones. Take a look at our HPLC Step by Step guide to understand chromatography in general.
-
- Protein Affinity: If you are lucky enough to buy resin with antibodies vs. your protein, you can simply pass your protein through the resin and it will selectively bind your protein. Then wash it a little bit with buffer so no other proteins are bound and finally elute it by disrupting the antibody-protein interaction.
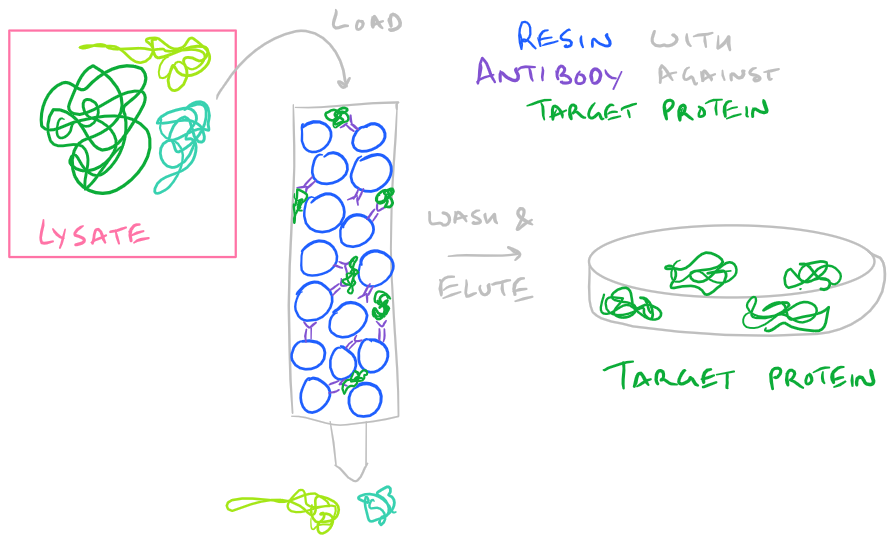

-
- Protein Substrate: If your protein is an enzyme with a binding pocket, you can also immobilize your substrate on a column and use that for purifying your protein. Simply pass your protein through the column multiple times so it binds the substrate while other non-functional proteins are easily washed away.
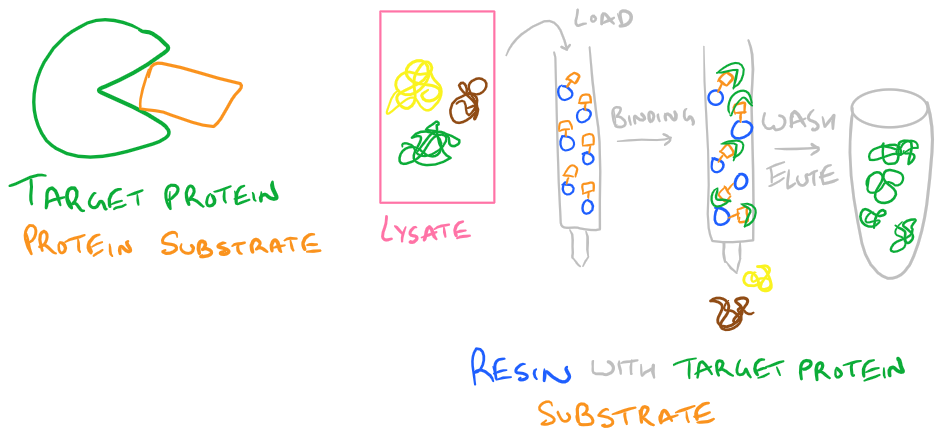
A typical protein purification strategy will involve using several of these techniques in combination. No single technique is 100% efficient, so each time you purify with one of these methods, your protein will get more and more pure. Use a western blot to analyze how clean your protein is. You can also use a silver stain to determine purity. I’ll discuss this technique in the future.
Note: Grammarly is a free grammar check plugin for Chrome. I used it for this article and really like it! Try it out here…
Purification of Recombinant Proteins with His Tags
Above, we have already discussed the purification of recombinant proteins via their charge and using their binding pocket. Another strategy that’s very popular is to introduce at least 6 Histidine residues into the N- or C-Terminus of a protein via cloning. Then, when it’s time for purification we can run the protein through a divalent nickel column. Histidine residues, at a high pH (~7.6), can chelate Nickel and hence will be bound on the nickel column. The column can be washed with a low concentration of Imidazole (~20 mM) and then eluted with 150 mM+ of Imidazole.
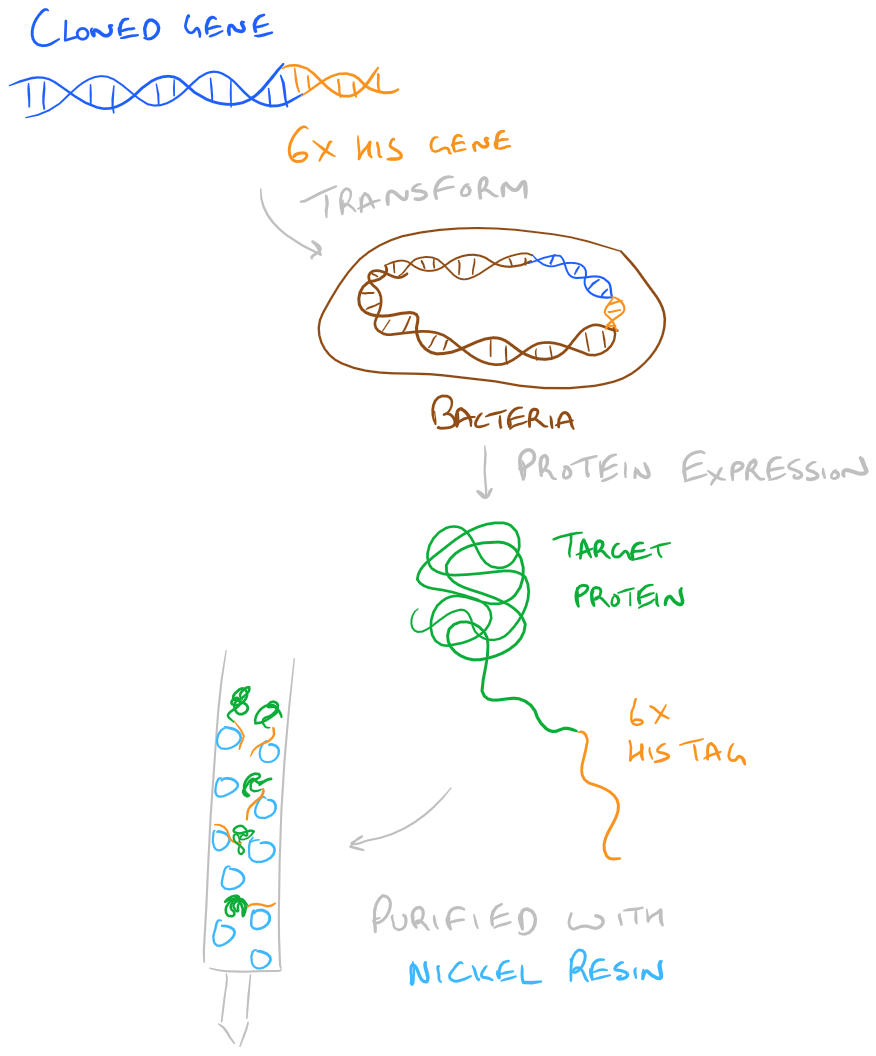
Step by Step Guide to Purification of a His-Tagged Fusion Protein
Materials
Neurospora culture
Lysis Buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 1 mM B-mercaptoethanol)
Homogenizer
Protease inhibitor cocktail
Phosphate buffered saline (pH 7.0)
Wash buffer (50 mM Phosphate Buffer pH 7.0, 300 mM NaCl, 1 mM Imidizole, pH 7.0 final)
Elution buffer (50 mM Phosphate Buffer pH 7.0, 300 mM NaCl, 150 mM Imidizole, pH 7.0 final)
Collection tubes for washes and elutions
Methods
- Grow culture and lyse in lysis buffer at 4 C for 45 min
- Homogenize lysate and centrifuge at 12000 g for 20 min
- Discard the pellet
- Dialyze the supernatant against PBS (pH 7.0) for 1 hour at 4 C. Replace the buffer outside the dialysis bag and continue to dialyze for 1 hour more.
- Prepare the Nickel-Agarose column according to the manufacturers instructions.
- Add in your protein dialysate from the previous steps on top of the column.
- Allow the material to diffuse to the bottom and load the filtrate on the column once again
- Wash the column with wash buffer (use 10x the volume of the beads in the column)
- Elute the column with elution buffer (use 1-3x the volume of the beads in the column)
- Collect the eluant in 1 ml fractions and assay each fraction for protein
- Assaying the protein can be performed via a western blot or other protein assay
Tips and Tricks for Purifying Recombinant Proteins with His Tags
- EDTA is used in lysis buffer to prevent protease activity
- Use a dialysis membrane of the appropriate size to retain your protein’s molecular weight + 1000 Da at least. This way you can be sure that you aren’t losing a lot of your protein along with all the filtrate.
- The size of the column that you use should be determined according to the instructions
- Protein assays for determining activity are a broad category. For many enzymes there are assays where the enzyme will be used to cleave a substrate and generate a fluorescent signal.
Protein Purification Protocols on Scigine
- 6x HIS Tag Purification of ATPase
- Purification of a His Tagged Fusion Protein
- Robotic purification via an affinity column
- Recombinant protein purified via Size Exclusion
- Metalloprotease Purification by Anion Exchange
Powerpoint related to various Purification Processes
References:
Guide to Protein Purification and Assays from NIH
Protein Purification Powerpoint Presentation
Applications of Protein Purification from Cornell
Manju Kapoor’s Guide to Protein Purification
Nickel-Agarose Purification Guide