Southern Blots: Overview & Theory
Southern Blotting is a technique that is used to detect whether you have a specific DNA sequence available in your sample. Most commonly, you will be testing for DNA that you get from cell lysates or after creating your own plasmids. You can also do some amazing things like making transgenic mice and proving that you have selected for certain genes of interest to you! All you have to do is Southern Blot any set of DNA from the mice and you’ll have concrete proof. As you probably understand, this is one of the most common techniques that geneticists and molecular biologists know because everything they do needs to be proved by a southern blot.
Note: If you’re writing research papers, I highly recommend Grammarly – it’s a free grammar check plugin for Chrome. Try it out here…
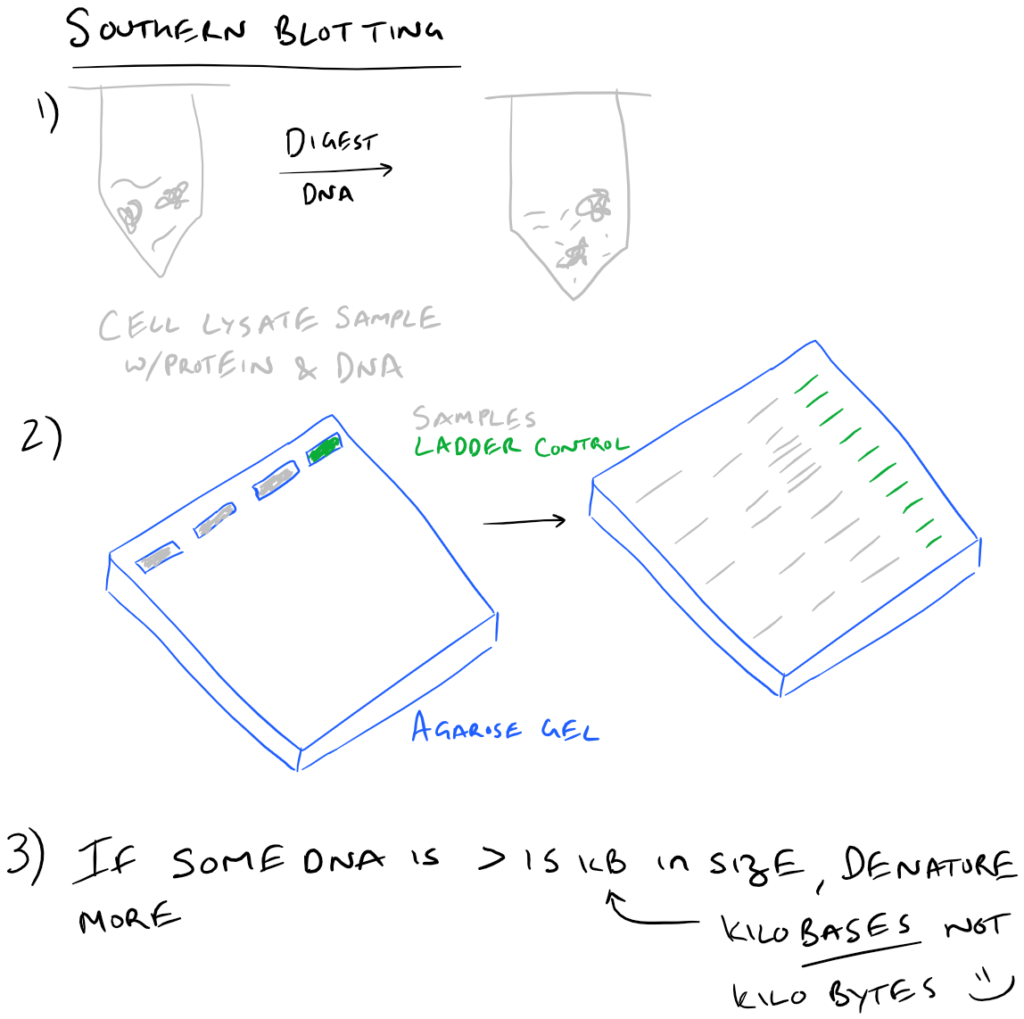
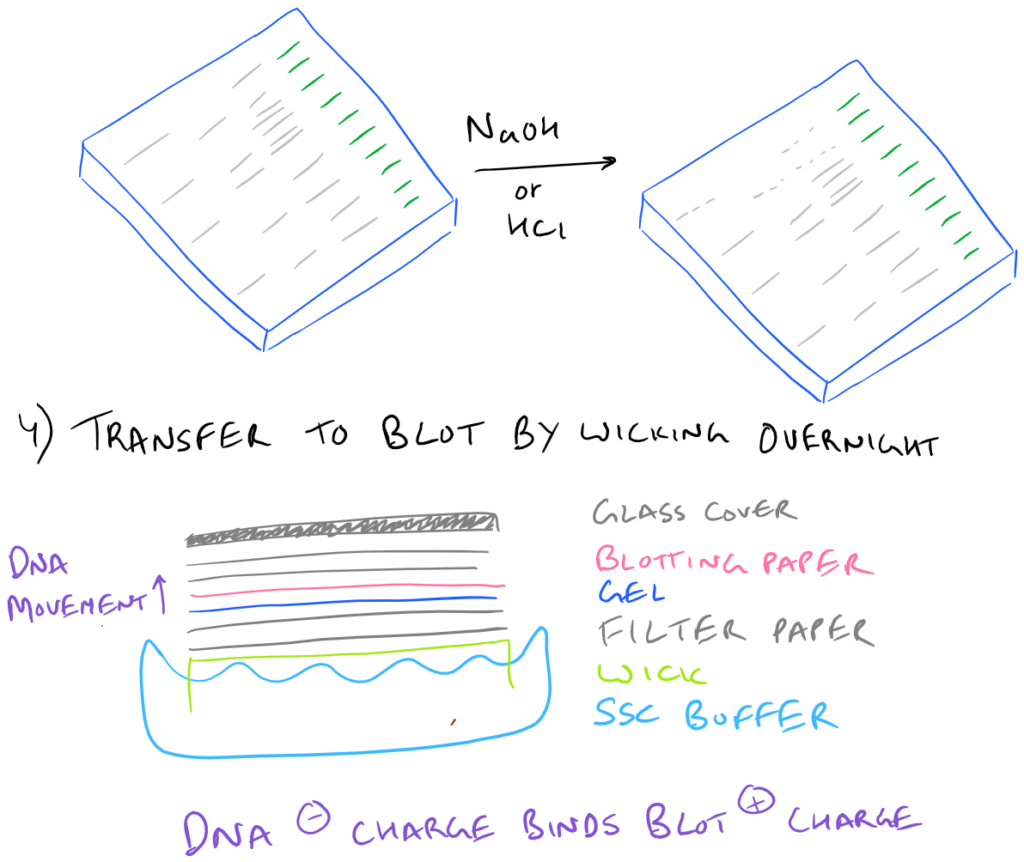
To execute a southern blot, first collect some DNA. This can be from a cell lysate or from tissue samples, etc. Next, digest the DNA using DNAse and run these fragments on an agarose gel. This will separate the DNA by size and you’ll know which well in the gel corresponds with which sample. Next, in a similar fashion to Western blots, this DNA is then transferred onto blotting paper (typically made of nitrocellulose or nylon). After the “blotting” process is complete, the DNA is then “probed” using a radioactive (or fluorescent) DNA sequence that is complementary to the sequence that you want to detect. Finally, this “probed” radioactive blot is then imaged using an autoradiograph. The steps for this process are described in the following image:
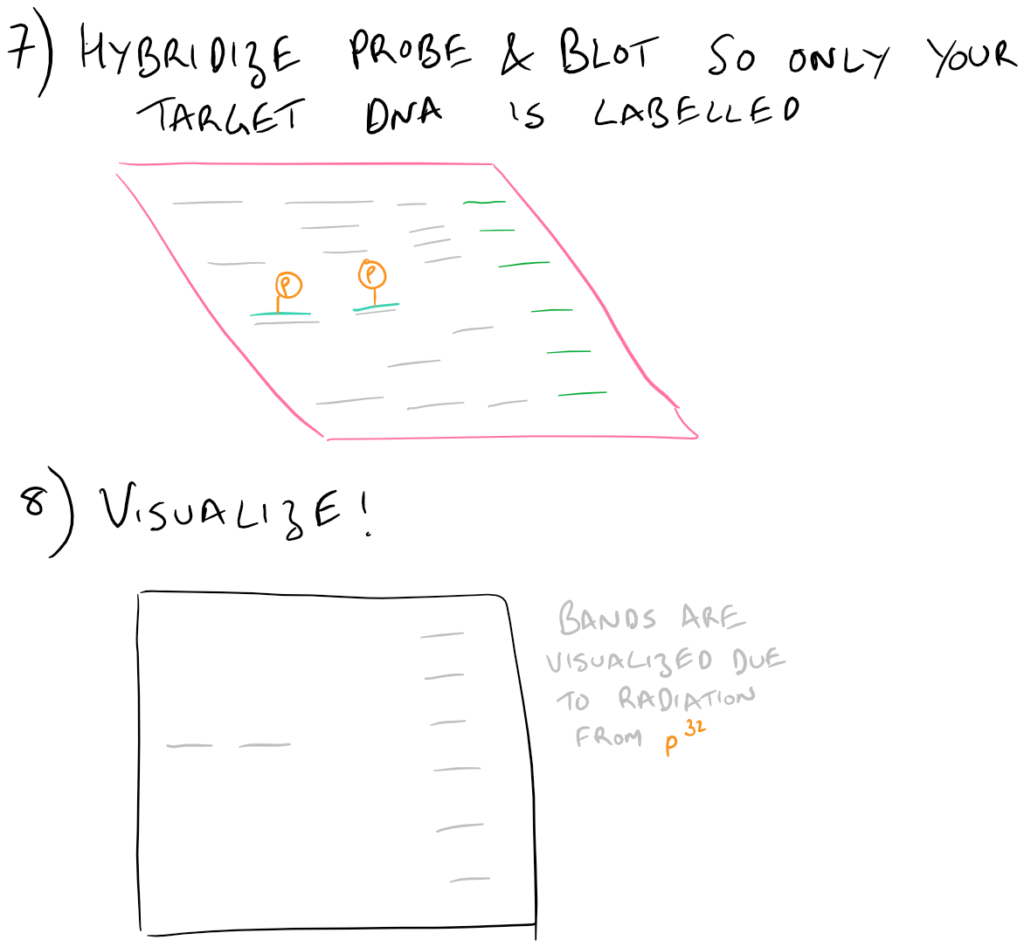
Nuances of Southern Blotting theory
Why is there is a second denaturing step for DNA that’s 15 KB or larger?
It’s because these large pieces of DNA don’t transfer very well to the blotting paper. As you can imagine, the larger a piece of DNA is, the slower it will migrate through an agarose gel. Even after leaving the blotting paper overnight, the transfer may not be complete!
How does the DNA actually go from the gel into the blot?
Through capillary action and wicking! Unlike a western blot where a voltage gradient is utilized to pull proteins into the blotting paper, in Southern Blots, the DNA merely moves over to the blotting paper overnight without much force at all. That’s why it is incredibly important to make sure that the blotting paper and the gel are in close contact. It’s also very important to make sure that the glass plate on top is heavy enough so that it forces the gel and the blot together.
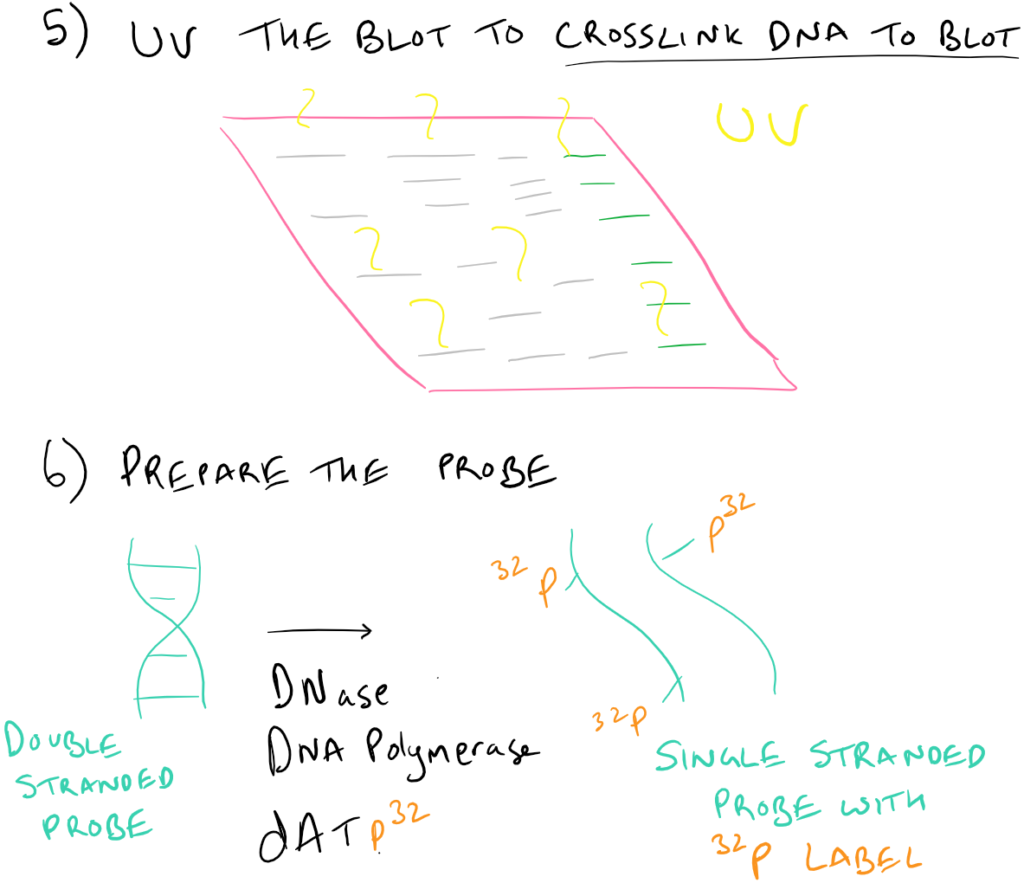
Why is there a UV step?
By using UV after the transfer step, the DNA (which has some free aldehyde groups due to depurination) can react with the nitrocellulose/nylon membrane to form covalent bonds.
What do the NaOH and the HCl do during the denaturation step?
Essentially HCl removes some/all of the purine bases from the DNA and makes the two DNA strands less sticky to each other (because there is less hydrogen bonding). This process is called depurination. NaOH also prevents the two strands from forming hydrogen bonds due to deprotonation of all bases.
Note: Grammarly is a free grammar check plugin for Chrome. I used it for this article and really like it! Try it out here…
How does radiolabeling with P32 work?
Small amounts of DNAse introduce nicks into the single stranded probe DNA. DNA polymerase then utilizes the dATP32 from solution to repair these nicks and incorporates these radioactive phosphates into the backbone of the DNA.
Southern blot Step-By-Step Guide
Materials for Southern Blot of Mouse Tail DNA
Denhardt’s Solution 50X (5g Ficoll 70000, 5g Polyvinyl pyrrolidone, 5g BSA Fraction V in 500 ml water)
Hybridization cocktail (25 ml 50% dextran sulfate, 25 ml 20X SSC, 50 ml formamide, 1 ml Tris 1M, 2 ml Denhardt’s Solution 50X, 1ml 10% SDS) – prefilter before use
TE buffer (10 mM Tris HCl pH 7.5, 1 mM EDTA)
BamHI enzyme and buffer (#R0136S New England BioLabs)
100X BSA (B9000S New England BioLabs)
Gel loading dye (#G2526 Sigma)
TBE buffer 10X (#93290 Sigma)
SSC buffer 20X (#S6639 Sigma)
[α32P]dCTP (3000 Ci/mmol; #NHG013H250UC Perkin Elmer)
Sephadex G-50 (#G50150 Sigma)
Ready-To-Go DNA Labeling Beads (-dCTP) (#27-9240-01 GE Lifesciences)
Whatman paper (#WHA10427810 Sigma)
Salmon Sperm DNA (#15632011 ThermoFisher)
Southern Blot Protocol
DNA digest and gel electrophoresis
- Setup DNA digest reactions from collected tail samples as follows:
10 µl DNA (12 ug)
6 µl 10X BamHI buffer
0.6 µl 100X BSA
1 µl BamHI
42.4 µl H2O
Incubate at 37°C for at least 5 hours.
- Add 6 µl gel loading dye to each sample.
- Run samples of a 0.8% agarose gel in TBE for 16 h at 25V.
- Stain gel in 1 µg/ml ethidium bromide for 30 min.
- Take a photo of the gel.*
Transfer
- Soak gel in 0.2 N HCl for 10 min with shaking to depurinate (remove purines from DNA). Rinse with water.
- Soak gel in two washes of 500 ml 0.4 M NaOH/1.5 M NaCl solution for 30 min each time.
- Soak gel in two washes of 500 ml 1 M Tris/1.5 M NaCl solution for 30 min each time.
- Setup transfer stack by placing 2 layers of Whatman paper on glass plate (wet before laying down) with soaked gel on top (flipped over). Cover with plastic wrap.
- Using a razor blade, cut out plastic over gel and place wetted membrane on gel. Place two layers of Whatman paper on top (wet before laying down).
- Place a stack of paper towels on the top and cover with a glass plate and transfer in 6X SSC overnight.
- Dismantle and air-dry membrane.
- Expose membrane to UV for 90 s (DNA side down). Bake between Whatman paper at 80°C for at least 1 h.
Labelling the probe
- Denature probe by adding 2 µl probe (25-50 ng) to 44 µl TE and incubating at 95°C for 4 min). Immediately place on ice.
- Add 46 µl denatured probe and 4 µl of [α32P]dCTP to Ready-to-Go labelling bead tube. Flick to mix.
- Incubate at 37°C for 15-30 min.
- Add 50 µl TE and run through a G50 sephadex column
Probe Hybridization and Autoradiograph
- Add 50 ml hybridization cocktail to the membrane.
- Add 300 µl of 10 mg/ml salmon sperm DNA to labelled probe and denature at 95°C for 5 min. Immediately place on ice. Add to membrane and cocktail solution.
- Hybridize overnight at 42°C.
- Wash briefly with 50 ml wash solution (0.2X SSC/0.1% SDS).
- Wash for 30 min with 100 ml wash solution at 50°C. Repeat.
- Check blot with Geiger counter to estimate cpm.
- Expose membrane to film and store at -70°C.
- Develop after 2-5 days.
Notes on this Southern Blot method
- During DNA digest Step 5 you can note the position of the marker bands by using a fluorescent ruler or marking bands directly on the gel by punching small holes with a needle.
- This method uses dCTP32 and not dATP32 as we talked about in the theory section
- After undergoing HCl and NaOH treatment for the denaturation step, it is very important to neutralize the gel once again.
- If you don’t want to buy the labelling beads, an alternative strategy is presented here at MIT Cores
Application of Southern Blots on SciGine, a Scientific Method Search Engine
References:
ASU Southern Blots Guide
MIT Core Guide
DNA Blot from Davidson College