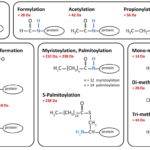
Ligate Sticky Ends via DNA Ligation
Ligation of Sticky Ends, Summary of DNA ligation We have already discussed a high level view of gene cloning in…
Protein Purification of Recombinant Proteins
Protein Purification Summary In our previous blog posts we have explored Gene cloning with Plasmid Vectors in Bacteria, Transient transfection…
Gene Cloning using Plasmids: Molecular Cloning Intro
Gene Cloning with Plasmids: Summary We all know that DNA is the basic building block of biology. So, how can…
Bacterial Transformation Protocol with Competent Cells
Bacterial Transformation using Competent Cells: Summary Since we have already learned Calcium Phosphate Transfection with mammalian cells, let’s now focus…
Guide: Measure Cell Proliferation with Thymidine and BrdU
Thymidine and BrdU, Cell Proliferation Assay Summary How do you know if your cultured cells are growing? Does your new…
Cytotoxicity & Cell Viability with MTT Assay Protocol
Cell Viability with MTT Assay Summary Cell Viability is a common technique used by biochemists who are studying oncology and…
Calcium Phosphate Transient Transfection Protocol & Guide
Transfection with Calcium Phosphate: General Summary Molecular biology tools allow us to understand and manipulate DNA/RNA so that we can…
RNA Extraction, Isolation, and Purification by SciGine
RNA Extraction: General Summary RNA extraction and isolation is a precursor for many methods in molecular biology including Northern Blotting,…
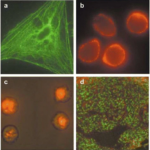
Immunofluorescence Microscopy Protocol and Method Guide
Immunofluorescence Microscopy Overview & Theory Immunofluorescence Microscopy (IF) is a classical technique to observe the localization of molecules in cell/tissue…
Immunoprecipitation (IP) Scientific Method Guide
Immunoprecipitation Overview Immunoprecipitation is a method for extracting protein from a solution. Typically, this solution is a cell lysate which…